# install
BiocManager::install("miloR")
# library
library(miloR)
library(SingleCellExperiment)
library(scater)
library(scran)
library(dplyr)
library(patchwork)miloR的学习与使用
miloR R包的使用
miloR Learning
1. 背景
John C. Marioni 教授是国际知名的计算生物学家,在位于英国剑桥希克斯顿(Hinxton)的惠康基因组研究园(Wellcome Genome Campus)长期担任重要领导职务。他的研究开创了单细胞基因组学数据分析的统计方法,对转录组学领域产生了深远影响。
miloR 在《Nature Biotechnology》是由John C. Marioni发表,Differential abundance testing on single-cell data using k-nearest neighbor graphs
R包 的链接 (https://github.com/MarioniLab/miloR)
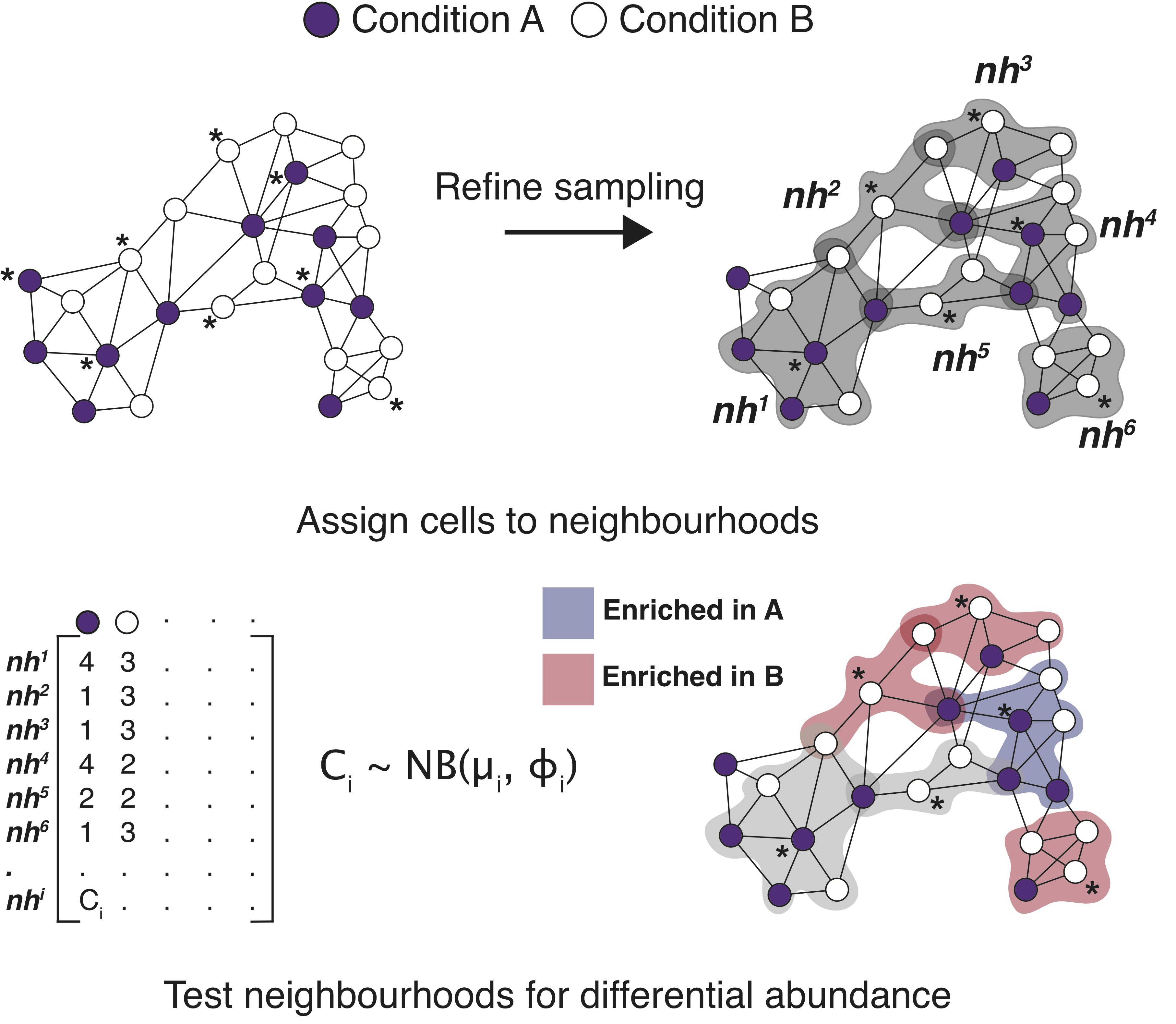
为了解决这些挑战,Marioni开发了一种计算框架,可以在不依赖将细胞聚类成离散组的情况下进行差异丰度测试。利用了一种常见于许多单细胞分析中的数据结构:k近邻(KNN)图。我们将细胞状态建模为该图上的重叠邻域,并将其作为差异丰度检验的基础。为了解决重叠邻域之间的非独立性问题,应用了加权版的Benjamini-Hochberg方法,其中P值由邻域连通性的倒数加权——这是对先前描述的用于控制空间错误发现率(FDR)策略的一种图式改进。
Note
Milo 的方法利用了广义线性模型 (GLM) 的灵活性,从而可以进行复杂的实验设计,例如纳入干扰和技术协变量,包括考虑从不同批次中确定的样本。
1.1 关键步骤
- 构建KNN图
基于降维后的细胞嵌入(如 PCA、UMAP),计算细胞间的欧氏距离,构建 K-最近邻图(KNN graph)。 每个细胞的邻域定义为与其最近的 k 个邻居(默认 k=30),形成重叠的局部细胞群体。
Milo 使用基于基因表达空间相似性计算的 KNN 图来表示细胞所在的表型流形。为了构建 KNN 图,我们遵循单细胞分析的最佳实践40,通过按每个细胞的测序深度重新缩放唯一分子标识符计数,应用对数变换,并将 M 个细胞的基因表达矩阵投影到 d 个主要主成分 (PC) 上。
原始数据中,每个细胞都有两万多个基因(维度)
- 降维后的细胞嵌入,把细胞放进坐标系
- PCA/UMAP的作用:将每个细胞简化为低维度空间中的一个点
- 空间含义:靠近的点表示转录组特征相似
- 构建KNN图:建立邻里关系
- KNN (K-Nearest Neighbors) 的核心是为每一个细胞找“邻居”
- 欧氏距离:算法计算一个细胞到其他所有细胞的直线距离
- 连线:对于细胞A,算法选出距离它最近的k个细胞(默认是30),并在它们之间连上一条线
- 结果:所有细胞和这些连线构成了一个巨大的社交网络,功能相似的细胞被紧密的锁在一个圈子里
- 定义领域(Neighborhoods)
对每个细胞,统计其所属的邻域(即作为其他细胞的邻居的次数),形成邻域-细胞矩阵。 通过 图抽样(graph sampling) 减少冗余,保留代表性邻域。
- Counting cells in neighborhoods
对于每个邻域,我们统计每个实验样本 S 中的细胞数量,构建一个 N × S(邻域 × 实验样本计数)矩阵。
- Testing for differential abundance in neighborhoods
对每个邻域中存在的细胞数量进行计数(每个实验样品),并将其用于条件之间的差异丰度测试。
为了检验差异丰度,我们使用 edgeR 中的 QL 方法分析邻域计数,类似于 Cydar 中的实现方式。
Tip
准似然检验(Quasi-Likelihood, QL)
QL 方法的优势在于它不仅估计了基因(或邻域)层面的离散度,还通过 QL 分散度(QL dispersion) 捕捉了数据中的不确定性,这比传统的似然比检验(Likelihood Ratio Test)更能有效地控制假阳性率(FDR)
我们对每个邻域的计数拟合一个负二项广义线性模型(NB GLM),并使用 TMM 归一化来考虑不同样本中细胞数量的差异,使用指定对比的 QL F 检验来计算每个邻域的 P 值。
- Controlling the spatial FDR in neighborhoods
使用 FDR(如 Benjamini-Hochberg)校正邻域水平的 p 值,控制假阳性率。
空间 FDR 可以解释为邻域并集中被假阳性邻域所占的比例。为了控制 KNN 图中的空间 FDR,我们应用了 Benjamini-Hochberg 方法的加权版本,其中 P 值由邻域连通性的倒数加权。作为邻域连通性的衡量标准,我们使用每个邻域中索引单元格到第 k 个最近邻的欧氏距离。
- 可视化
为了可视化邻域差异分析的结果,我们构建了一个抽象图,其中节点代表邻域,边代表邻域间共有的细胞数量。节点的大小代表邻域内的细胞数量。节点的位置由采样索引细胞在单细胞均匀流形近似和投影(UMAP)中的位置决定,以便与单细胞嵌入进行定性比较。图形可视化使用 R 包 ggplot 和 ggraph 实现。
2. miloR Tutorials
2.1 Basic Milo 模拟数据
- 加载数据
使用miloR自带的数据集,traj_sce 是大的SingleCellExperiment数据类型
Tip
之前已经介绍过 seurat ,是当前最常用的单细胞转录组分析工具,主要是从原始数据到聚类注释再到可视化,是一个高度封装、一站式解决方案,但是seurat背后的数据结构–SingleCellExperiment(SCE)
SingleCellExperiment 是 Bioconductor 项目设计的基础类,继承自 SummarizedExperiment,专为单细胞数据存储与分析而优化。SCE 可以统一存储表达矩阵、细胞/基因元数据、降维结果等信息,并被上百个 Bioconductor 单细胞工具所支持。通过 Seurat v5 中的 as.SingleCellExperiment() 与 as.Seurat() 函数,可以在两个体系之间自由转换。
“想象 SCE 对象是一艘货轮,每个 slot 就是一个专用货箱,分别存储不同类型的数据,整船出发就是一次完整的数据分析旅程
data("sim_trajectory", package = "miloR")
## Extract SingleCellExperiment object
traj_sce <- sim_trajectory[['SCE']]
## Extract sample metadata to use for testing
traj_meta <- sim_trajectory[["meta"]]
## Add metadata to colData slot
colData(traj_sce) <- DataFrame(traj_meta)
colnames(traj_sce) <- colData(traj_sce)$cell_id
redim <- reducedDim(traj_sce, "PCA")
dimnames(redim) <- list(colnames(traj_sce), paste0("PC", c(1:50)))
reducedDim(traj_sce, "PCA") <- redim 看完SingleCellExperiment 之后,就能了解到数据集中数据的组成和含义了
> traj_sce
class: SingleCellExperiment
dim: 500 500
metadata(0):
assays(2): counts logcounts
rownames(500): G1 G2 ... G499 G500
rowData names(0):
colnames(500): C1 C2 ... C499 C500
colData names(2): cell_id group_id
reducedDimNames(1): PCA
mainExpName: NULL
altExpNames(0):- rowData 表示基因信息
- colData 表示细胞信息
- assays 表示count信息
- reducedDimNames 表示降维信息
- Pre-processing
对于DA分析,需要构建单细胞的无向KNN图,标准的单细胞分析流程通常基于PCA中的距离来实现这一点,使用scater进行归一化并计算主成分还可以运行UMAP进行可视化
logcounts(traj_sce) <- log(counts(traj_sce)+1)
traj_sce <- runPCA(traj_sce, ncomponents = 30)
traj_sce <- runUMAP(traj_sce)
plotUMAP(traj_sce)- Create a Milo object
用不同来源的数据,构建Milo数据结构,核心还是获取到细胞基因的表达矩阵
(1). From SingleCellExperiment object
为了对图邻域进行差异丰度分析,首先构建一个 Milo 对象。该对象扩展了 SingleCellExperiment 类,用于存储 KNN 图上邻域的信息。
# 3. create milo
traj_milo <- Milo(traj_sce)
reducedDim(traj_milo, "UMAP") <- reducedDim(traj_sce, "UMAP")Milo是SCE的延续,增加了新的slot用来记录后续的数据
> traj_milo
class: Milo
dim: 500 500
metadata(0):
assays(2): counts logcounts
rownames(500): G1 G2 ... G499 G500
rowData names(0):
colnames(500): C1 C2 ... C499 C500
colData names(5): cell_id group_id Condition Replicate Sample
reducedDimNames(2): PCA UMAP
mainExpName: NULL
altExpNames(0):
nhoods dimensions(2): 1 1
nhoodCounts dimensions(2): 1 1
nhoodDistances dimension(1): 0
graph names(0):
nhoodIndex names(1): 0
nhoodExpression dimension(2): 1 1
nhoodReducedDim names(0):
nhoodGraph names(0):
nhoodAdjacency dimension(2): 1 1nhoods:这是最关键的矩阵。它记录了哪些细胞属于哪个邻域- 通常是一个二元矩阵(0 或 1),行代表细胞,列代表邻域中心。
nhoodIndex:存储了作为每个邻域“圆心”(Index cells)的细胞索引。Milo 并不会在每个细胞周围都建邻域,而是挑选一部分具有代表性的细胞作为中心nhoodCounts: 邻域丰度矩阵- 行是邻域,列是样本
- 微分丰度(Differential Abundance)分析的基础数据
- nhoodExpression: 记录了每个邻域中基因的平均表达水平。这对于后续分析“哪个邻域对应哪种细胞特征”非常有用
nhoodDistances: 记录了邻域中心细胞之间的距离nhoodAdjacency: 邻域的邻接矩阵,记录了哪些邻域之间是有重叠或相邻关系的nhoodGraph/graph: 存储了基于邻域构建的图结构
(2). From AnnData object (.h5ad)
可以使用 zellkonverter 包,从存储为 h5ad 文件的 AnnData 对象创建一个 SingleCellExperiment 对象。
library(zellkonverter)
# Obtaining an example H5AD file.
example_h5ad <- system.file("extdata", "krumsiek11.h5ad",
package = "zellkonverter")
example_h5ad_sce <- readH5AD(example_h5ad)
example_h5ad_milo <- Milo(example_h5ad_sce)(3). From Seurat object
从Seurat转换也是常见的方法,但是必须是SingleCellExperiment
library(Seurat)
data("pbmc_small")
pbmc_small_sce <- as.SingleCellExperiment(pbmc_small)
pbmc_small_milo <- Milo(pbmc_small_sce)- Construct KNN graph
将 KNN 图添加到 Milo 对象中。该图以 igraph 格式存储在 graph 插槽中。miloR 包包含从存储在 reducedDim 插槽中的 PCA 维度构建和存储该图的功能
traj_milo <- buildGraph(traj_milo, k = 10, d = 30)结果是一个复杂的igraph对象,其核心数据结构主要由两部分组成
- 节点(Vetices):数量等于数据中细胞总数,每个节点都有唯一的ID
- 边(Edges): AB连成一条线
> traj_milo@graph
$graph
IGRAPH 3d3d461 U--- 500 3920 --
+ edges from 3d3d461:
[1] 1-- 4 1-- 11 1-- 18 1-- 38 1-- 78 1-- 92 1--155 1--164 1--184 1--194 1--195 1--243 1--260 1--274
[15] 1--280 1--358 1--392 1--405 1--436 1--456 1--486 1--499 2-- 95 2--187 2--198 2--202 2--226 2--233
[29] 2--322 2--393 2--478 2--482 3-- 5 3-- 20 3-- 43 3-- 57 3--153 3--290 3--299 3--300 3--333 3--491
[43] 4-- 92 4-- 99 4--214 4--270 4--298 4--304 4--328 4--358 4--366 4--401 4--468 4--469 5-- 20 5-- 24
[57] 5-- 43 5-- 49 5-- 51 5-- 54 5-- 57 5-- 75 5-- 86 5-- 88 5--108 5--112 5--133 5--162 5--204 5--215
[71] 5--254 5--259 5--290 5--296 5--299 5--318 5--333 5--367 5--395 5--407 5--451 5--466 5--474 5--491
[85] 6-- 80 6--115 6--136 6--143 6--199 6--204 6--263 6--269 6--275 6--286 6--318 6--330 6--333 6--466
[99] 7-- 28 7-- 56 7--104 7--111 7--134 7--285 7--398 7--416 7--435 7--465 8-- 17 8-- 40 8-- 78 8-- 92
[113] 8-- 98 8--104 8--134 8--155 8--288 8--304 8--366 8--416 8--469 9-- 27 9-- 44 9-- 46 9-- 51 9-- 54
+ ... omitted several edges- IGRAPH UN– [节点数] [边数] –
- UN (Undirected, Unweighted)
- 界定代表性neighbourhoods
上一步中将单元格的邻域(索引)定义为 KNN 图中与索引单元格通过边连接的单元格组。不测试每个细胞邻域内的 DA,对代表性细胞子集进行采样作为索引
prop:随机抽样的初始细胞比例(通常 0.1 - 0.2 就足够了)k: 用于 KNN 细化的 k 值(建议使用与 KNN 图构建相同的 k 值)d: 用于 KNN 细化的降维维度数量(建议使用与 KNN 图构建相同的 d 值)refined指明是否要使用采样细化算法,或者只是随机选择单元格,默认且推荐的方法是使用优化算法。只有当您使用基于图的校正算法(例如 BBKNN)对数据进行批量校正后,DA 测试结果仍不理想时,才可以考虑使用随机算法
定义好邻域之后,最好查看一下邻域的大小(即每个邻域包含多少个单元格)。这会影响DA测试的效力。
traj_milo <- makeNhoods(traj_milo, prop = 0.1, k = 10, d=30, refined = TRUE)
plotNhoodSizeHist(traj_milo)dgCMatrix类,d:代表 double。表示矩阵中存储的是双精度浮点数(实数);g:代表 general。表示这是一个一般的矩阵,没有特殊的对称性或三角结构;C代表 column-compressed。表示它是按列压缩存储的。
traj_milo@nhoods
- 行(Rows):代表所有的细胞(与原始输入数据中的细胞一一对应)
- 列(Columns):代表被选为邻域中心的索引细胞(Index Cells)
- 数值:
- 1: 表示该行对应的细胞属于该列对应的邻域
- 0: 表示该行对应的细胞不属于该邻域
- 计算邻域cells
需要统计每个样本中每个邻域内的细胞数量,需要使用细胞元数据,并指定哪一列包含样本信息。
traj_milo <- countCells(traj_milo, meta.data = data.frame(colData(traj_milo)), samples="Sample")这会在 Milo 对象中添加一个 n × m 矩阵,其中 n 是邻域的数量,m 是实验样本的数量。 m 的值表示每个样本在每个邻域中计数的细胞数量。该计数矩阵将用于 DA 测试。
Note
colData是要取细胞对应的信息包括细胞信息,样本信息,分组信息,疾病信息等
核心是输入样本信息的列
- Differential abundance testing
广义线性模型 (GLM) 框架中实现了这一假设检验,主要是
edgeR中的Negative Binomial GLM
首先需要考虑实验设计。设计矩阵应该将样本与感兴趣的条件相匹配。在本例中,条件就是我们要检验的协变量。
traj_design <- data.frame(colData(traj_milo))[,c("Sample","Condition")]
traj_design <- distinct(traj_design)
rownames(traj_design)<- traj_design$Sample
traj_design <- traj_design[colnames(nhoodCounts(traj_milo)), , drop=FALSE]Milo 采用了 cydar 引入的空间 FDR 校正的改进版本,该版本考虑了邻域之间的重叠。具体来说,每个假设检验的 P 值都按第 k 个最近邻距离的倒数进行加权。要使用此统计数据,首先需要将最近邻居之间的距离存储在 Milo 对象中。
traj_milo <- calcNhoodDistance(traj_milo, d=30)
rownames(traj_design) <- traj_design$Sample
da_results <- testNhoods(traj_milo, design = ~ Condition, design.df = traj_design)- 可视化 为了可视化 DA 结果,我们构建了一个抽象的邻域图,我们可以将其叠加到单细胞嵌入上
plotUMAP(traj_milo) + plotNhoodGraphDA(traj_milo, da_results, alpha=0.05) +
plot_layout(guides="collect")2.2 利用 Milo 进行差异丰度检验——小鼠原肠胚形成示例
For this vignette we will use the mouse gastrulation single-cell data from Pijuan-Sala et al. 2019. The dataset can be downloaded as a SingleCellExperiment object from the MouseGastrulationData package on Bioconductor. To make computations faster, here we will download just a subset of samples, 4 samples at stage E7 and 4 samples at stage E7.5.
该数据集已经过预处理,并包含一个经过 pca 校正的降维,该降维是在使用 fastMNN 进行批量校正后构建的
# 0. Load packages
library(miloR)
library(SingleCellExperiment)
library(scater)
library(scran)
library(dplyr)
library(patchwork)
library(MouseGastrulationData)
# 1. Loda data
select_samples <- c(2, 3, 6, 15,
# 4, 19,
10, 14, 20, 30
#31, 32
)
embryo_data = EmbryoAtlasData(samples = select_samples)
# 2. Visualize the data
embryo_data <- embryo_data[,apply(reducedDim(embryo_data, "pca.corrected"), 1, function(x) !all(is.na(x)))]
embryo_data <- runUMAP(embryo_data, dimred = "pca.corrected", name = 'umap')
plotReducedDim(embryo_data, colour_by="stage", dimred = "umap")
# 3. DA testing
## 3.1 Create a milo object
embryo_milo <- Milo(embryo_data) %>%
buildGraph(embryo_milo, k = 30, d = 30, reduced.dim = "pca.corrected")%>%
makeNhoods(embryo_milo, prop = 0.1, k = 30, d=30, refined = TRUE, reduced_dims = "pca.corrected")
## 3.2 average neighbourhood size over 5 x N_samples.
plotNhoodSizeHist(embryo_milo)
## 3.3 Counting cells in neighbourhoods
embryo_milo <- countCells(embryo_milo, meta.data = as.data.frame(colData(embryo_milo)), sample="sample")
## 3.4 Defining experimental design
embryo_design <- data.frame(colData(embryo_milo))[,c("sample", "stage", "sequencing.batch")]
embryo_design$sequencing.batch <- as.factor(embryo_design$sequencing.batch)
embryo_design <- distinct(embryo_design)
rownames(embryo_design) <- embryo_design$sample
## 3.5 Computing neighbourhood connectivity
embryo_milo <- calcNhoodDistance(embryo_milo, d=30, reduced.dim = "pca.corrected")
## 3.6 Testing
da_results <- testNhoods(embryo_milo, design = ~ sequencing.batch + stage, design.df = embryo_design)
## 3.7 Inspecting DA testing results
ggplot(da_results, aes(PValue)) + geom_histogram(bins=50)
ggplot(da_results, aes(logFC, -log10(SpatialFDR))) +
geom_point() +
geom_hline(yintercept = 1) ## Mark significance threshold (10% FDR)
embryo_milo <- buildNhoodGraph(embryo_milo)
### Plot single-cell UMAP
umap_pl <- plotReducedDim(embryo_milo, dimred = "umap", colour_by="stage", text_by = "celltype",
text_size = 3, point_size=0.5) +
guides(fill="none")
### Plot neighbourhood graph
nh_graph_pl <- plotNhoodGraphDA(embryo_milo, da_results, layout="umap",alpha=0.1)
umap_pl + nh_graph_pl +
plot_layout(guides="collect")
plotDAbeeswarm(da_results, group.by = "celltype")
Note
不健康的分布(\(k\) 太小):直方图在左侧(接近 0 的地方)有一个很高的峰。这说明你的邻域太“碎”了,每个邻域包含的细胞不足以支撑后续的统计检验。
不健康的分布(\(k\) 太大):虽然统计效力提高了,但邻域会变得过于庞大,导致不同细胞类型的特征被混在一起,失去了空间分辨率(即你无法区分到底是哪种微小的细胞状态发生了变化)。