devtools::install_github("jinworks/CellChat")CellChat了解与使用
CellChat V2版本的使用
CellChat Learning
1. 细胞通讯分析(CellChat)背景
Tip
细胞通讯是指一个信号产生细胞发出的信息通过介质(即配体)传递到另一个靶细胞并与其相应的受体相互作用,然后通过细胞信号转导产生靶细胞内一系列生理生化变化,最终表现为靶细胞整体的生物学效应的过程。
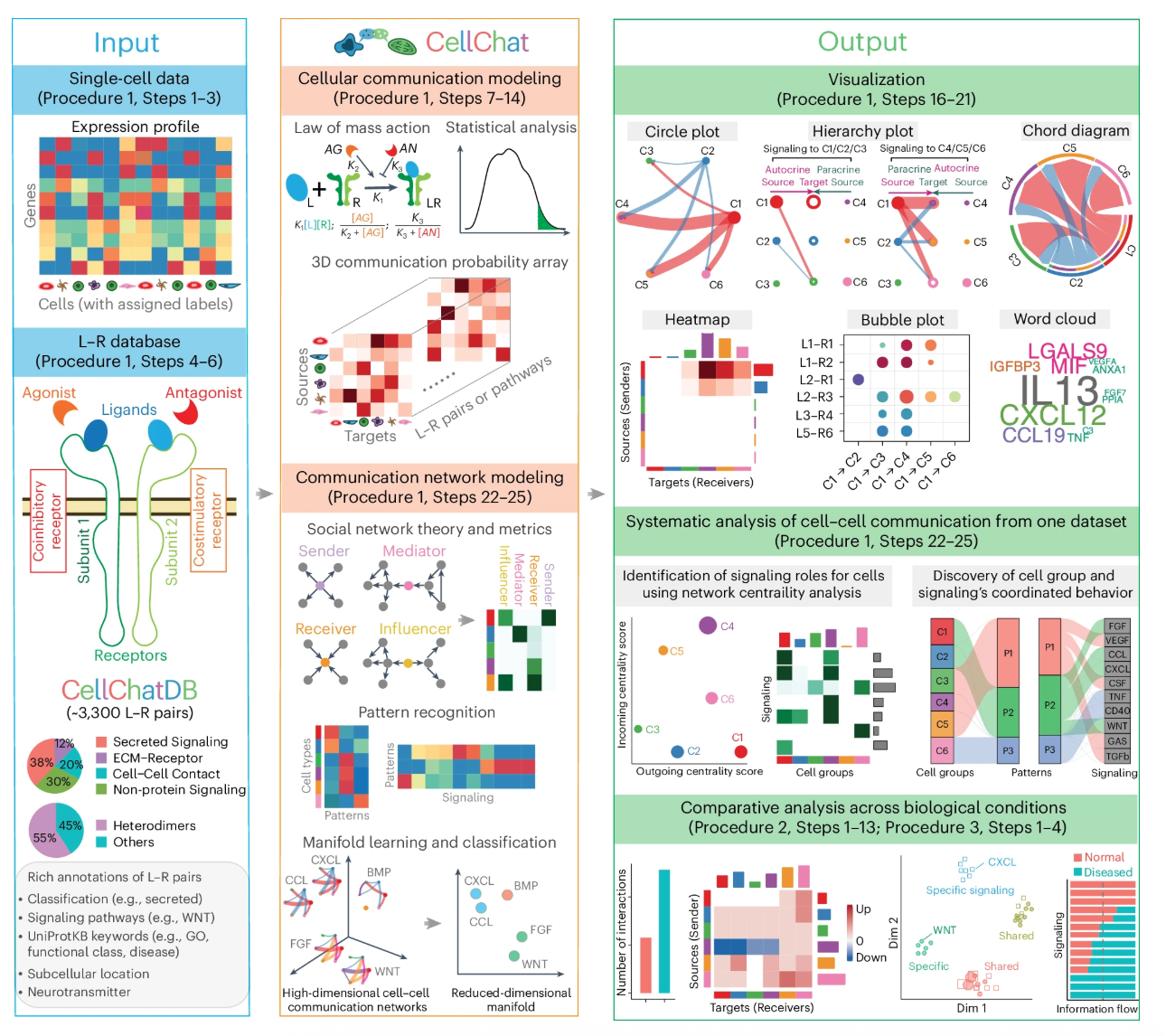
1.1 细胞间相互作用
在生物体中,不同细胞类型和组织中的细胞之间相互作用(CCI)可以协调生物的发育。
CCI通过各种分子包括离子、代谢物、整联蛋白、受体、连接蛋白、结构蛋白、配体和细胞外基质的分泌蛋白来实现的。一些分子如激素、生长因子、趋化因子、细胞因子和神经递质等配体介导细胞之间的信息交流
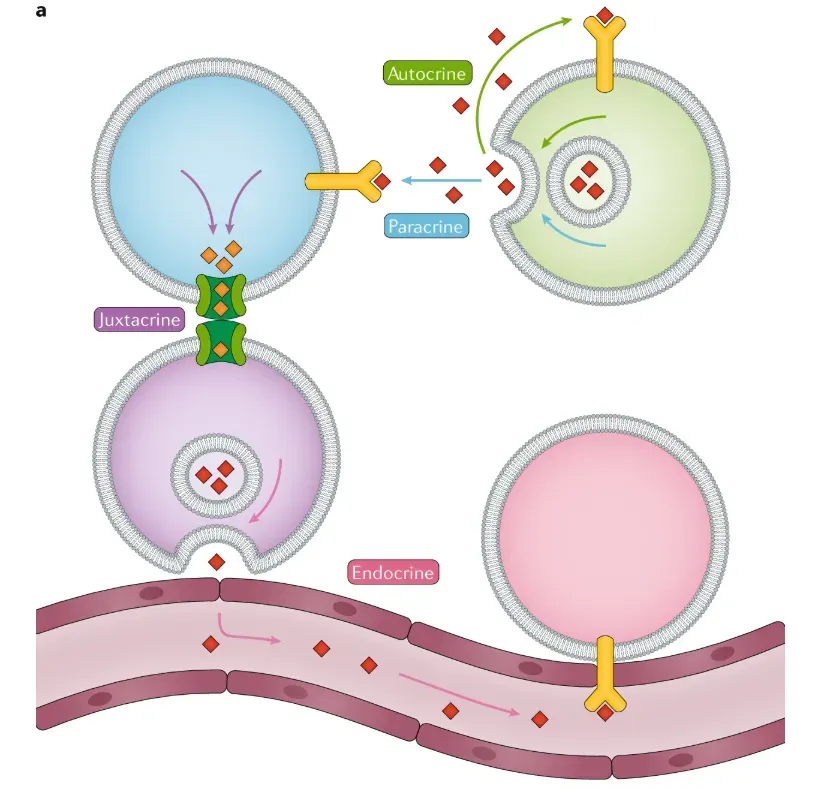
1.2 细胞通讯类型
自分泌信号转导是指细胞内通讯,细胞分泌配体,这些配体用于通过同源受体诱导同一细胞上表达的那些分子的细胞应答。
旁分泌细胞间的通讯不需要细胞间的接触,而是取决于信号分子在分泌后从一个细胞扩散到另一个细胞
近分泌,即依赖于接触的细胞间通讯依赖于间隙连接或膜纳米管等其他结构,使信号分子直接在细胞之间传递,而不会分泌到细胞外
内分泌细胞间的通讯代表细胞间的通讯,信号分子被分泌并通过诸如血浆的细胞外液传播很长一段距离
细胞间通讯背后的信号转导通常是由各种类型的蛋白质相互作用介导的。包括配体-受体,受体-受体和细胞外基质-受体的相互作用。
受体细胞通过同源受体触发下游信号传导,通常最终导致转录因子活性和基因表达发生改变,从而使这些表达发生改变的细胞进一步与其微环境相互作用。
2. CellChat
单细胞转录组-cellchat细胞通讯
CellChat优势
- 考虑多种subunits/ cofactors/ 聚合物 - 附带注释database并支持可视化结果图 - 支持物种为人或者小鼠
CellChat细胞通讯数据库通过整合KEGG和文献,构建了细胞通讯数据库cellchatDB,数据库主要包括三部分 - 旁分泌、自分泌的信号互作 - 胞外基质-受体互作 - 细胞直接互作 - 除了直接互作的蛋白外,cellchat还将可溶性激动剂、拮抗剂等互作引子考虑在内
2.1 细胞通讯原理
Note
根据细胞表达的配体(Ligand)和受体(Receptor)推断细胞之间是否存在信号传递,并量化其强度。
细胞通讯的可能性通过配受体基因表达量计算,如果配体是复合物,则取表达量的几何平均数,如果受体是复合物,则取表达量的几何平均数。对于受体配体的复合物来说,只要其中一个亚基的表达量为0,则认为这对受体配体对不起作用。
细胞通讯显著性计算采用置换检验来检验每个受体配体的显著性。置换检验的思路为随机置换细胞类群的标签,以此标签计算特定配体受体对的通讯可能性。置换的次数默认为100次,比较置换后的特定配体受体对通讯的可能性和真实的通讯可能性,计算显著性p值。
- 构建Ligand-Receptor 知识库
细胞不能直接说话,必须通过 - 配体(Ligand): 由发送细胞分泌(CXCL12.TGFB1.WNT5A) - 受体(Receptor): 由接收细胞表达(CXCR4,TGFBR1,FZD)
CellChat内置大型人工整理数据库
- 配体-受体二元对(CXCL-CXCR)
- 配体复合物
- 受体复合物
- 多组分信号通路
- 细胞外基质相互作用
- 根据单细胞表达确定”哪些细胞能产生信号”
- 配体的平均表达量
- 受体的平均表达量
- 是否高于表达背景
- 是否满足配体/受体复合体的构建要求
只有显著表达配体的细胞才能作为信号发送端(Sender) 只有显著表达受体的细胞才能作为信号接收端(Receiver)
- 计算每一对细胞之间的信号强度
通讯概率(communication probability)= 配体表达 × 受体表达 × 细胞数量校正 × 通路权重
- 多配体、多受体子单元的最小表达限制
- 结构蛋白复合体的组装概率
- 细胞数量差异校正(避免“大群体压倒一切”的误差)
- 构建完整的细胞通讯网络进行可视化
- 环形图(Circle plot):展示不同细胞之间的通路连接
- 层级图(Hierarchy plot):展示信号的“发送–接收方向性”
- 热图(Heatmap):比较不同细胞间通讯强度
- 气泡图(Bubble plot):展示特定配体–受体在 sender/receiver 的表达组合
- 中心性分析(Centrality analysis):判断哪些细胞是网络中心
- 多样本对比
- 通路富集分析
- 细胞通讯强度变化分析
- 信号网络“主导者”(sender)、“目标”(receiver)、“调节者”(moderator)的识别
| 信号通路类别 | 代表通路(CellChat 中常见) | 主要参与细胞类型 | 核心生物学功能 | 常见研究场景 |
|---|---|---|---|---|
| 趋化与免疫迁移信号 | CXCL、CCL | T 细胞、B 细胞、巨噬细胞、树突状细胞 | 调控免疫细胞迁移、组织定位、炎症反应 | 肿瘤免疫微环境、炎症、自身免疫 |
| 炎症与免疫调节信号 | MIF、TNF、IL1 | 巨噬细胞、单核细胞、肿瘤细胞 | 介导炎症反应、免疫激活或免疫抑制 | 肿瘤、慢性炎症、感染 |
| 免疫检查点 / 抑制信号 | TGF-β、PD-1/PD-L1(部分) | T 细胞、Treg、肿瘤细胞 | 抑制免疫应答、诱导免疫耐受 | 肿瘤免疫逃逸 |
| 生长与分化信号 | WNT、FGF、EGF | 上皮细胞、干细胞、成纤维细胞 | 细胞增殖、分化、干性维持 | 发育生物学、肿瘤发生 |
| 细胞命运决定信号 | NOTCH | 干细胞、免疫细胞 | 决定细胞命运、分化方向 | 发育、免疫分化、肿瘤干性 |
| 血管生成相关信号 | VEGF、ANGPT | 内皮细胞、成纤维细胞 | 血管生成、血管稳定性 | 肿瘤血管、组织修复 |
| 基质与细胞黏附信号 | COLLAGEN、LAMININ、FN1 | 成纤维细胞、上皮细胞、肿瘤细胞 | 细胞黏附、迁移、组织结构维持 | 肿瘤侵袭、纤维化 |
| 整合素介导信号 | INTEGRIN | 成纤维细胞、免疫细胞 | 细胞-基质相互作用、机械信号感知 | ECM 重塑、肿瘤转移 |
| 代谢与应激相关信号 | IGF、HGF | 肿瘤细胞、基质细胞 | 代谢调控、细胞存活 | 肿瘤代谢、组织损伤 |
| 神经-免疫相关信号 | NRG、SEMA | 神经细胞、免疫细胞 | 神经调控、免疫调节 | 神经系统、肿瘤神经浸润 |
| 发育与形态建成信号 | BMP、HEDGEHOG | 干细胞、间充质细胞 | 器官发育、形态建成 | 胚胎发育、再生医学 |
2.2 CellChat 安装
CellChat 在2024年更新到了V2版本,能够
- 从空间分辨转录组学推断相互作用细胞群之间空间邻近的细胞间通讯
- CellChatDB v2 数据库进行了扩展,新增了 1000 多个蛋白质和非蛋白质相互作用(例如代谢和突触信号传导),并附有丰富的注释。此外,还提供了一个名为 updateCellChatDB 的函数,方便用户轻松更新 CellChatDB。
安装最近的CellChat 可以直接在R中使用devtools进行安装
如果网络不行,需要在github中下载 CellChat.zip 文件,然后使用 devtools::install_local安装,但是必须安装对应的依赖包才行
Install NMF (>= 0.23.0)usinginstall.packages('NMF')Install circlize(>= 0.4.12)Install ComplexHeatmap- Install UMAP python pacakge for dimension reduction:
pip install umap-learn
2.3 单个数据集的CellChat完整流程
CellChat官网上有最详细的教程,其他都是看了之后进行再学习的,千人千见,可以自行学习 单数据学习
- Data input & processing and initialization of CellChat object
1.1 Data Load
使用Seurat自带的数据集进行,或者自己的数据集进行

# install data package
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("satijalab/seurat-data")
library(infb.SeuratData)
library(SeuratData)
InstallData("ifnb")
library(seurat)
# load data
library(ifnb.SeuratData)
data("ifnb")
ifnb <- UpdateSeuratObject(ifnb)
table(ifnb$orig.ident)- 从
Seuratobject 导入数据
data输入使用的是normalized data matrix,所以要根据Seurat版本来决定data的数据层
# seurat V4 data load
data.input <- seurat_object[["RNA"]]@data
# seurat V5 data load
data.input <- GetAssayData(scemerge_sub, assay = "RNA", layer = "data")
# lables
labels <- Idents(seurat_object)
meta <- data.frame(labels = lables, rown.names = names(lables))- 从
SingleCellExperimentobject获取data
data.input <- SingleCellExperiment::logcounts(object)
meta <- as.data.frame(SingleCellExperiment::colData(object))
meta$labels <- meta[["sce.clusters"]]1.2 Create a CellChat object
cellchat can create a new CellChat object from a data matrix, Seurat, SingleCellExperiment or AnnData object,对象中的元数据将默认使用,用户必须提供 group.by 来定义单元格组。例如,group.by = “ident” 表示 Seurat 对象中的默认单元格标识。
CellChat可以从归一化的data创建对象,常规的方法,但是需要补充meta文件,以及设置group.by,当然也提供了从Seurat,SingleCellExperiment,AnnData直接构建cellchat
- Starting from the digital gene expression matrix and cell label information
cellchat <- createCellChat(object = data.input, meta = meta, group.by = "labels")- Starting from Seurat object
# change Idents
Idents(cellchat) <- "CellType"
# create cellchat
cellChat <- createCellChat(object = seurat.obj, group.by = "ident", assay = "RNA")- Starting from SingleCellExperiment object*
cellChat <- createCellChat(object = sce, group.by = group.by = "sce.clusters")当使用原始数据进行构建cellchat object时候,可以用使用自带的函数
cellchat <- addMeta(cellchat, meta = meta)
cellchat <- setIdent(cellchat, ident.use = "labels") # set "labels" as default cell identity
levels(cellchat@idents) # show factor levels of the cell labels
groupSize <- as.numeric(table(cellchat@idents)) # number of cells in each cell group1.3 Set the ligand-receptor interaction database
在用户能够使用 CellChat 推断细胞间通讯之前,他们需要设置配体-受体相互作用数据库,并识别过度表达的配体或受体。CellChatDB v2 包含约 3300 个经过验证的分子相互作用,其中包括约 40% 的分泌自分泌/旁分泌信号相互作用、约 17% 的细胞外基质 (ECM)-受体相互作用、约 13% 的细胞间接触相互作用以及约 30% 的非蛋白质信号传导。与 CellChatDB v1 相比,CellChatDB v2 新增了 1000 多个蛋白质和非蛋白质相互作用,例如代谢和突触信号传导。
CellChat v2 利用配体和受体的关键介质或酶来估计配体和受体的表达,从而预测非蛋白质介导的潜在通讯。
CellChatDB <- CellChatDB.human # use CellChatDB.mouse if running on mouse data
showDatabaseCategory(CellChatDB)
# Show the structure of the database
dplyr::glimpse(CellChatDB$interaction)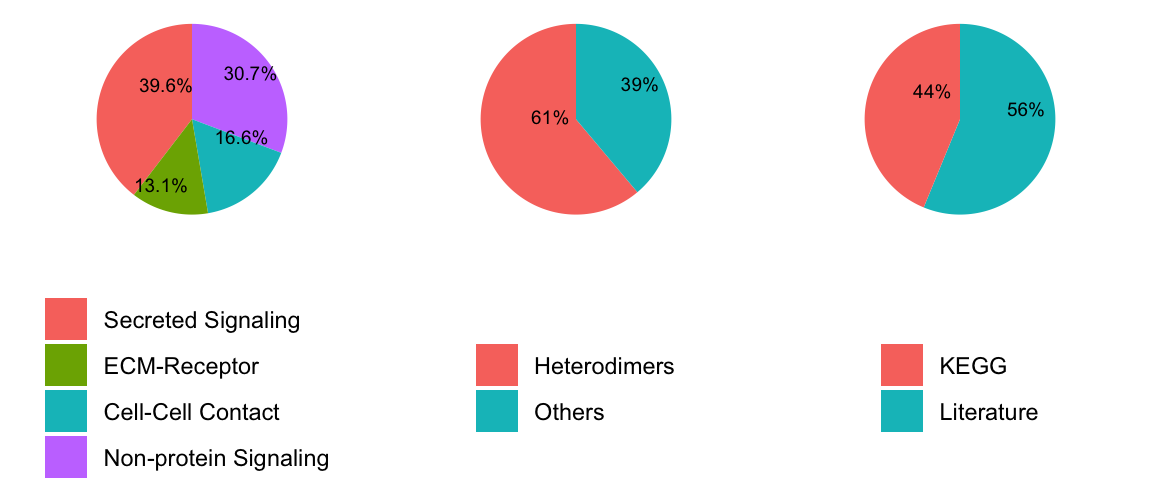
选择自己想依据的数据库,然后把想用的数 据库替换掉cellchat@DB
# use a subset of CellChatDB for cell-cell communication analysis
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling", key = "annotation") # use Secreted Signaling
# Only uses the Secreted Signaling from CellChatDB v1
CellChatDB.use <- subsetDB(CellChatDB, search = list(c("Secreted Signaling"), c("CellChatDB v1")), key = c("annotation", "version"))
# use all CellChatDB except for "Non-protein Signaling" for cell-cell communication analysis
CellChatDB.use <- subsetDB(CellChatDB)
# set the used database in the object
cellchat@DB <- CellChatDB.use1.4 细胞通讯数据预处理
为了推断细胞状态特异性通讯,CellChat 识别一个细胞群中过度表达的配体或受体,然后如果配体或受体过度表达,则识别过度表达的配体-受体相互作用。
提供一个将基因表达数据投影到蛋白质-蛋白质相互作用(PPI)网络上的功能,采用扩散过程,根据基因邻近基因的表达值来平滑基因的表达值,这些邻近基因的表达值是在高置信度的实验验证的蛋白质-蛋白质网络中定义的。当分析测序深度较浅的单细胞数据时,此功能非常有用,因为投影可以减少信号基因的丢失效应,特别是对于配体/受体亚基可能出现的零表达。
library(CellChat)
# subset the expression data of signaling genes for saving computation cost
# This step is necessary even if using the whole database
cellchat <- subsetData(cellchat)
future::plan("multiprocess", workers = 4)
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
# # #> The number of highly variable ligand-receptor pairs used for signaling inference is 692
# project gene expression data onto PPI (Optional: when running it, USER should set `raw.use = FALSE` in the function `computeCommunProb()` in order to use the projected data)
# cellchat <- projectData(cellchat, PPI.human)通过蛋白质-蛋白质相互作用(PPI)网络来优化单细胞数据,从而更准确地推断细胞间的通讯(如配体-受体相互作用)
逻辑:它参考了一个经过实验验证的、高置信度的 PPI 网络
如果基因 A 在某个细胞里表达量为 0,但它在 PPI 网络中的“邻居基因”表达量很高,算法就会根据邻居的表现,给基因 A “补充”一点数值(平滑处理)
Note
默认情况下,CellChat是不使用衍射,如果要启动
- 计算通讯概率之前,先进行投影函数
projectData(cellchat, PPI.human)
- 计算通讯概率之前,先进行投影函数
- 在计算通讯概率的时候,设置
computeCommunProb(object, raw.use = FALSE)
- 在计算通讯概率的时候,设置
- Inference of cell-cell communication network
2.1 Compute the communication probability and infer cellular communication network
CellChat 通过给每次相互作用赋予一个概率值并进行置换检验来推断具有生物学意义的细胞间通讯。CellChat 利用质量作用定律,将基因表达与信号配体、受体及其辅助因子之间相互作用的先验知识相结合,从而模拟细胞间通讯的概率。
在化学中,质量作用定律是指:化学反应的速率与反应物浓度的乘积成正比
- 配体(Ligands): 发出的信号
- 受体(Receptors): 接收的开关
- 辅助因子(Auxiliary factors): 有些信号需要“帮手”才能生效
- 激动剂:增强信号
- 拮抗剂:屏蔽信号
Important
先验知识的作用(Prior Konwledge) 内置数据库
trimean方法: 统计学上非常Robust方法,结合了中位数和四分位数 \[ Trimean = \frac{Q_1 + 2M + Q_3}{4} \]
如果研究的生物过程中众所周知的信号通路没有被预测出来,用户可以尝试使用较低的 trim 值来改变计算每个细胞组平均基因表达的方法。
# 耗时
cellChat <- computeCommunProb(cellChat, type = "triMean")- To use 10% truncated mean, USER can set type = “truncatedMean” and trim = 0.1
- To determine a proper value of trim, CellChat provides a function computeAveExpr, which can help to check the average expression of signaling genes of interest
2.2 将推断出的蜂窝通信网络提取为数据框
我们提供了一个名为 subsetCommunication 的函数,以便轻松访问感兴趣的推断细胞间通讯。
df.net <- subsetCommunication(cellchat)returns a data frame consisting of all the inferred cell-cell communications at the level of ligands/receptors. Set slot.name = “netP” to access the the inferred communications at the level of signaling pathwaysdf.net <- subsetCommunication(cellchat, sources.use = c(1,2), targets.use = c(4,5))gives the inferred cell-cell communications sending from cell groups 1 and 2 to cell groups 4 and 5.df.net <- subsetCommunication(cellchat, signaling = c("WNT", "TGFb"))gives the inferred cell-cell communications mediated by signaling WNT and TGFb.
2.3 Infer the cell-cell communication at a signaling pathway level
CellChat 通过汇总与每个信号通路相关的所有配体-受体相互作用的通信概率,计算信号通路层面的通信概率。每个配体-受体对和每个信号通路的推断细胞间通讯网络分别存储在“net”和“netP”槽中。
cellchat <- computeCommunProbPathway(cellchat)2.4 计算聚合的细胞间通信网络
CellChat 通过计算链路数量或汇总通信概率来计算聚合的小区-小区通信网络。用户还可以通过设置 sources.use 和 targets.use 来计算细胞组子集中的聚合网络。
cellchat <- aggregateNet(cellchat)CellChat can also visualize the aggregated cell-cell communication network. For example, showing the number of interactions or the total interaction strength (weights) between any two cell groups using circle plot.
CellChat也带有可视化的函数
最基础的就是细胞间的网络环状通讯图
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = groupSize, weight.scale = T, label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize, weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")由于细胞间通讯网络的复杂性,我们可以检测每个细胞群发送的信号。此外,我们还控制了参数 edge.weight.max,以便比较不同网络之间的边权重。
mat <- cellchat@net$weight
par(mfrow = c(3,4), xpd=TRUE)
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(mat2, vertex.weight = groupSize, weight.scale = T, edge.weight.max = max(mat), title.name = rownames(mat)[i])
}- Visualization of cell-cell communication network
在推断出细胞间通信网络后,CellChat 提供了多种功能,用于进一步的数据探索、分析和可视化。
- hierarchical plot, circle plot, Chord diagram, and bubble plot.
- 分离和可视化高顺位的细胞网络推断信息
3.1 Visualize each signaling pathway using Hierarchy plot, Circle plot or Chord diagram
Hierarchy plot: 层级图/分层图 (用来展示“层级关系(父–子关系)或嵌套结构”的可视化图形,强调从整体到局部的组织结构,而不是数值变化趋势) 用户应定义 vertex.receiver,这是一个数值向量,表示层次结构图左侧目标单元格组的索引。该层级图由两部分组成:左侧部分显示了对特定目标细胞群(即定义的顶点接收器)的自分泌和旁分泌信号,右侧部分显示了对数据集中其余细胞群的自分泌和旁分泌信号。
不是基因编号、不是细胞数量,而是 CellChat 里 cell group 的编号 vertex.receiver = “谁是信号的接收方(receiver)”
Chord diagram: 弦图, cellchat 提供了netVisual_chord_cell 和 netVisual_chord_gene 可视化不同目的和水平的细胞通讯,netVisual_chord_cell用来可视化不同细胞组之间的细胞通讯,netVisual_chord_gene 用来可视化多种受体-配体的细胞通讯,用于可视化由多个配体-受体或信号通路介导的细胞间通讯(其中弦图中的每个扇形代表一个配体、受体或信号通路)
Explnations of edge color/weight, node color/size/shape: 在所有可视化图中,边的颜色与发送者的来源一致,边的权重与交互强度成正比。
Visualization of cell-cell communication at different levels:可以使用 netVisual_aggregate 可视化推断的信号通路通信网络,并使用 netVisual_individual 可视化与该信号通路相关的单个 L-R 对的推断通信网络。